






Gli inibitori di integrasi hanno rivoluzionato il trattamento dell’infezione da HIV grazie alla loro elevata potenza e tollerabilità. L’integrasi catalizza il trasferimento del DNA virale a doppio filamento, generato dalla trascrizione inversa del genoma, nel DNA della cellula ospite. Questa operazione richiede il taglio di due nucleotidi alle estremità 3’ del DNA virale e il trasferimento in una regione specifica del DNA della cellula ospite, anch’essa tagliata dallo stesso enzima. Dall’inibizione di quest’ultimo passaggio, deriva la denominazione della classe degli inibitori di integrasi finora entrati in uso clinico (integrase strand transfer inhibitors, INSTI), con la prima (raltegravir, elvitegravir) e la seconda generazione (dolutegravir, DTG; bictegravir e cabotegravir, CAB) che costituisce la base della maggioranza degli odierni regimi antiretrovirali di combinazione.
Nonostante gli INSTI di seconda generazione siano farmaci potenti e ad elevata barriera genetica, la terapia prolungata, spesso associata ad una aderenza subottimale, ad interazioni farmacologiche e alla presenza di popolazioni virali resistenti selezionate da terapie precedenti, può favorire l’emergenza di mutazioni che causano il fallimento della terapia.
Il meccanismo più comune di resistenza è associato alla selezione di mutazioni chiave nel gene dell’integrasi, che ne alterano il legame con il farmaco pur conservando la funzione enzimatica. Tuttavia, come già descritto per gli inibitori di proteasi (1,2), le mutazioni di resistenza possono emergere anche al di fuori del gene che codifica per la proteina bersaglio del farmaco (meccanismo epistatico). La resistenza epistatica nei confronti degli INSTI è stata osservata sia nel tratto polipurinico localizzato all’estremità terminale 3’ del genoma virale (3’-PPT) sia in alcune regioni codificanti per l’envelope e per il nucleocapside virale (NC).
Mutazioni nel gene dell’integrasi
Le mutazioni che più comunemente causano la riduzione di suscettibilità agli INSTI di prima generazione coinvolgono principalmente alcune posizioni amminoacidiche chiave (66, 118, 138, 140, 148 e 155). L’impiego degli INSTI di seconda generazione ha ridotto notevolmente questa evenienza, pur definendo qualche nuova mutazione (es. R263K). Considerata la rarità della selezione di queste mutazioni con gli INSTI di seconda generazione (3), la diminuzione o perdita di attività di questi farmaci riguarda soprattutto i casi con pregresso fallimento con INSTI di prima generazione poiché molte delle mutazioni resistenza da questi selezionate risultano essere cross-resistenti nei confronti degli INSTI di seconda generazione.
CAB si distingue dagli INSTI di seconda generazione per la sua bassa barriera genetica, con insorgenza di resistenza nella maggioranza dei fallimenti virologici (4). Resta da capire il peso relativo delle componenti di questa diversità: barriera ridotta intrinseca, uso in duplice terapia con farmaco a bassa barriera oppure farmacocinetica della formulazione esclusivamente long-acting iniettiva.
Mutazioni nel tratto polipurinico
L’attività degli INSTI blocca l’integrazione del DNA provirale nel genoma umano; tuttavia, quest’ultimo persiste come DNA non integrato che si riduce progressivamente in presenza di terapia efficace. Il DNA lineare è la forma più abbondante di DNA virale non integrato (60-80%), seguita dalle forme circolari contenenti 1 (20-30%) oppure 2 (2-5%) copie della regione di regolazione LTR (long terminal repeat).
Recentemente è stata documentata la possibilità di resistenza a DTG sia in vitro (5-7) che in vivo (8) in conseguenza di mutazioni emergenti in un breve tratto di 15 basi puriniche localizzato nella parte terminale dell’RNA virale, tra il gene nef e l’estremità 3’ della regione U3 della LTR, indicato come 3’-PPT, che serve come sito di innesco per la sintesi del filamento positivo di DNA durante la retrotrascrizione del genoma virale.
Le mutazioni selezionate in vitro in seguito a pressione farmacologica con DTG non hanno un pattern mutazionale univoco ma condividono la capacità di ridurre o abolire completamente le funzioni del 3’-PPT. Questa perdita di attività limita la produzione di DNA virale lineare e favorisce l’accumulo di forme non integrate circolari a 1-LTR (Figura 1) che supportano l’espressione di proteine virali e la generazione di particelle virali (5).
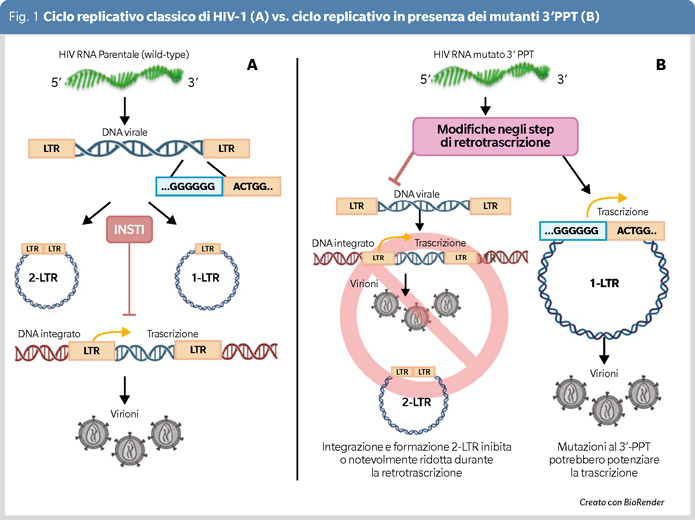
Uno dei profili mutazionali più rilevanti selezionato in vitro (GGGGGG→GCAGT-delezione) ha una capacità replicativa ridotta rispetto al virus privo di mutazioni di circa 90 volte, ma è resistente nei confronti di DTG circa 23 volte. Nel trial clinico DOMONO, caratterizzato dalla somministrazione di DTG in monoterapia (8), il fallimento virologico in assenza di mutazioni chiave nel gene dell’integrasi è stato riscontrato in un individuo che aveva sviluppato un pattern mutazionale peculiare al 3’-PPT (GGGGGG→GGGAGC); tale profilo, tuttavia, non si è confermato resistente in vitro (6).
Per chiarire l’impatto clinico delle mutazioni al 3’-PPT è necessaria un’analisi sistematica dei casi di fallimento dei regimi basati su INSTI in assenza di mutazioni nella regione bersaglio, unitamente a controlli sull’associazione del fallimento con la somministrazione di altre terapie. Attualmente non ci sono evidenze che questo fenomeno sia causa frequente di resistenza agli INSTI in assenza di mutazioni chiave nel gene bersaglio.
Mutazioni nel nucleocapside e nell’envelope virale
In due studi recenti (9-10), esperimenti di selezione di resistenza in vitro con DTG hanno rivelato la comparsa di mutazioni nella regione che codifica per la proteina del NC e nel gene env, in assenza di mutazioni primarie nel gene dell’integrasi ed indipendentemente dal tropismo corecettoriale del virus.
Le mutazioni nel NC conferiscono una resistenza modesta nei confronti del DTG (circa 4 volte) mentre quelle in env possono aumentare la resistenza fino ad oltre 1000 volte nel caso del mutante che include le mutazioni V85A, S162K, R298K, Q363R nella gp120, le mutazioni A541V, V693I, G825E nella gp41 e la mutazione K6N nella proteina Vpu. È interessante notare che questa variante risulta debolmente resistente anche nei confronti di altre classi di antiretrovirali, configurando quindi un meccanismo generale che però ha impatto soprattutto sugli INSTI (Figura 2).
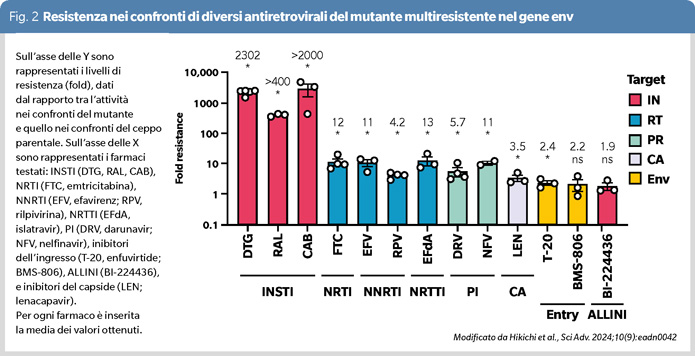
Ma con quale meccanismo le mutazioni off-target generano resistenza? E perché la resistenza emergente impatta soprattutto sul trattamento con INSTI? Per rispondere a queste domande dobbiamo considerare che le proteine gp120 e gp41 sono coinvolte nel legame con i recettori e corecettori presenti sulle cellule bersaglio e con i successivi processi di fusione fra envelope virale e membrana cellulare. Inoltre, l’infezione da HIV può essere sia di tipo extracellulare, cioè mediata dai virioni rilasciati dalle cellule infette, sia cellulo-mediata, ossia veicolata dal contatto diretto tra una cellula infetta e una non infetta. Le mutazioni in env associate alla resistenza agli INSTI, emergono nell’interfaccia tra la gp120 e la gp41, ne alterano la glicosilazione e la stabilità conformazionale, riducendo la capacità fusogena dell’envelope virale. Ciò favorisce l’infezione cellulo-mediata e comporta l’ingresso di un maggior numero di virioni generando un numero più alto di infezioni produttive.
All’aumento del rapporto tra virioni e cellule cresce anche il numero di intasomi, i complessi costituiti da DNA virale, integrasi e fattori cellulari necessari per l’integrazione del DNA virale nel genoma cellulare. L’attività degli INSTI diminuirebbe in conseguenza di un fenomeno di amplificazione del bersaglio poiché anche un singolo intasoma non bloccato dal farmaco può avviare un’infezione produttiva. Al contrario altri inibitori, come ad esempio gli inibitori della trascrittasi inversa, hanno più opportunità di bloccare l’infezione poiché il processo di retrotrascrizione richiede molti eventi catalitici.
Questo meccanismo di resistenza non sembra tuttavia avere impatto sugli inibitori di integrasi di natura allosterica (ALLINI), nuova classe in via di sviluppo che agisce più tardivamente rispetto agli INSTI, inducendo l’aggregazione delle molecole di integrasi ed ostacolando l’assemblaggio del genoma virale con le proteine strutturali.
Conclusioni
Gli INSTI di ultima generazione sono la classe regina della terapia antiretrovirale, grazie alla loro potenza e tollerabilità e alla barriera genetica elevata. Tuttavia, fenomeni inattesi di resistenza possono derivare da mutazioni fuori dalla regione codificante l’integrasi. Questa eventualità è stata finora dimostrata in vitro ma solo occasionalmente in vivo (limitatamente al meccanismo basato sulle mutazioni in 3’-PPT).
È auspicabile che l’impiego di metodiche di sequenziamento dell’intero genoma virale, attualmente in sviluppo, consenta in un prossimo futuro di definire in modo compiuto il ruolo delle mutazioni epistatiche nella risposta del virus ai trattamenti antiretrovirali.
- Castain L, Perrier M, Charpentier C, et al. New mechanisms of resistance in virological failure to protease inhibitors: Selection of non-described protease, Gag and Gp41 mutations. J. Antimicrob. Chemother. 2019;74:2019-2023.
- Coetzer M, Ledingham L, Diero L, et al. Gp41 and Gag amino acids linked to HIV-1 protease inhibitor-based second-line failure in HIV-1 subtype A from Western Kenya. J. Int. AIDS Soc. 2017;20;e25024.
- Buzon-Martin L, Navarro-San Francisco C, Fernandez-Regueras M, et al. Integrase strand transfer inhibitor resistance mediated by R263K plus E157Q in a patient with HIV infection treated with bictegravir/tenofovir alafenamide/emtricitabine: case report and review of the literature. J. Antimicrob. Chemother. 2024;79;1153-1156.
- Rusconi S, Santoro MM, Capetti AF, et al. The future of long-acting cabotegravir plus rilpivirine therapy: deeds and misconceptions. Int. J. Antimicrob. Agents. 2022;60(3):106627.
- Dekker JG, Klaver B, Berkhout B, et al. HIV-1 3’-Polypurine Tract Mutations Confer Dolutegravir Resistance by Switching to an Integration-Independent Replication Mechanism via 1-LTR Circles. J Virol. 2023;97(5):e0036123.
- Hachiya A, Kubota M, Shigemi U, et al. Specific mutations in the HIV-1 G-tract of the 3’-polypurine tract cause resistance to integrase strand transfer inhibitors. J Antimicrob Chemother. 2021;77(3):574-577.
- Richetta C, Subra F, Malet I, et al. Mutations in the 3’-PPT Lead to HIV-1 Replication without Integration. J Virol. 2022;96(14):e0067622.
- Wijting IEA, Lungu C, Rijnders BJA, et al. HIV-1 Resistance Dynamics in Patients with Virologic Failure to Dolutegravir Maintenance Monotherapy. J Infect Dis. 2018;218(5):688-697.
- Hikichi Y, Van Duyne R, Pham P, et al. Mechanistic Analysis of the Broad Antiretroviral Resistance Conferred by HIV-1 Envelope Glycoprotein Mutations. mBio. 2021;12(1):e03134-20.
- Hikichi Y, Grover JR, Schäfer A, et al. Epistatic pathways can drive HIV-1 escape from integrase strand transfer inhibitors. Sci Adv. 2024;10(9):eadn0042.
