






La colangite biliare primitiva (CBP), già nota come cirrosi biliare primitiva, è una malattia cronica del fegato che interessa più frequentemente il sesso femminile, con decorso generalmente molto lento. Se non diagnosticata e trattata, progredisce verso la cirrosi (CE), l’epatocarcinoma (HCC) e l’insufficienza d’organo.
I sintomi caratteristici della CBP, prurito e intensa stanchezza, sono presenti in oltre la metà dei casi sin dall’esordio clinico, tuttavia i pazienti vengono spesso sottoposti ad esami diagnostici e valutazioni specialistiche (dermatologiche, reumatologiche) che ritardano la diagnosi.
La disponibilità di terapie efficaci, soprattutto negli stadi non avanzati, impone una corretta conoscenza di questa malattia che, per quanto rara, ha un forte impatto sociale ed economico.
Epidemiologia
L’epidemiologia della CBP, classicamente considerata una malattia tipica delle donne in età media, è cambiata nel corso degli ultimi decenni. La CBP è ancora oggi più frequente nel sesso femminile, ma il rapporto femmine:maschi si è ridotto da 9:1 a 4-6:1. L’incidenza globale stimata è di 1.76 e la prevalenza di 14.6 per 100.000 abitanti (1).
I dati di un recente studio condotto sui database di 900 Medici di Medicina Generale indicano che in Italia l’incidenza è di 5.31 per 100.000/anno e la prevalenza di 27.9/100.000 (2). La CBP è più frequente nei paesi del Nord America e Nord Europa, anche se incidenza e prevalenza sembrerebbero in aumento in tutto il mondo. Nel sesso maschile la CBP tende ad avere una maggior severità alla diagnosi e una prognosi peggiore, con maggior rischio di HCC.
La diagnosi è più frequente tra la quarta/sesta decade di vita, ma in anni più recenti si assiste a diagnosi in età più avanzata e con malattia più lieve.
A conferma del ruolo patogenetico di fattori genetici, la CBP è molto più frequente in parenti di primo grado di pazienti con CBP. Diversi fattori ambientali (inquinamento atmosferico, fumo di sigarette, smalto per unghie e tinture per capelli, infezioni delle vie urinarie) sembrerebbero aumentare il rischio di CBP.
Diagnosi e inquadramento clinico
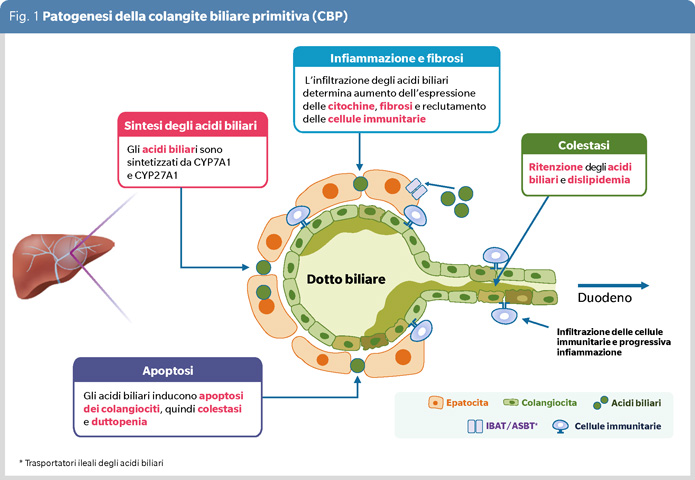
La CBP riconosce una patogenesi autoimmunitaria, al pari della epatite autoimmune (AIH) e della colangite sclerosante primitiva (CSP). Vi è consenso nel ritenere che in individui geneticamente predisposti l’azione di fattori ambientali determina la perdita di tolleranza immunitaria delle cellule dell’epitelio dei piccoli e medi dotti biliari, come dimostra la presenza di anticorpi anti-mitocondrio (AMA) in >95% dei pazienti. L’attivazione dei processi infiammatori determina colestasi e fibrosi, che nelle fasi più avanzate può causare duttopenia (scomparsa dei dotti biliari nei tratti portali) e cirrosi (Figura 1).
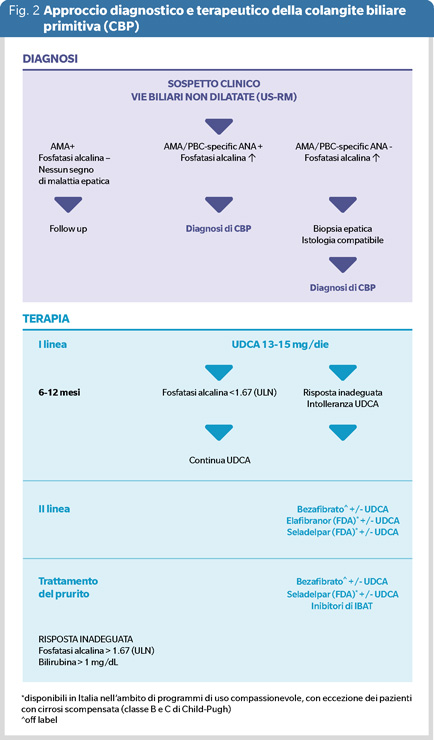
La diagnosi si basa sulla presenza di almeno due tra i seguenti criteri:
- fosfatasi alcalina (ALP) sopra la norma;
- positività sierologica per gli anticorpi anti-mitocondrio (AMA), e nei pazienti AMA negativi, presenza degli anticorpi anti-nucleo (ANA) CBP-specifici, sp100 e gp210
- segni istologici caratteristici (colangite cronica non suppurativa, epatite dell’interfaccia, danno dei piccoli e medi dotti biliari, duttopenia) (3).
L’ultrasonografia e la Risonanza Magnetica sono utili per escludere l’ostruzione delle vie biliari o la diagnosi di CSP. In corso di CBP i valori di GGT aumentano parallelamente alla ALP, ma in alcuni casi con ALP normale, la GGT può essere l’unico test biochimico precoce di malattia. Le IgM sono aumentate e rappresentano un marcatore utile per la diagnosi.
Alcuni rari pazienti con positività isolata per AMA, senza segni di danno epatico, possono nel tempo sviluppare la malattia, e pertanto necessitano di monitoraggio annuale.
La biopsia epatica è indicata solo nei pazienti con ALP nella norma e/o negatività auto-anticorpale, e nei pazienti con sospetto di AIH o di altre cause di colestasi cronica. Un incremento delle transaminasi >5 volte la norma deve indurre al sospetto di overlap con epatite autoimmune (CBP-AIH) o di variante autoimmune. Resta controverso se considerare questi rari casi come due condizioni sovrapposte o una variante della CBP.
In alcuni pazienti con maggior espressione autoimmunitaria è indicato associare un farmaco immunosoppressore.
La biopsia epatica riveste oggi un ruolo limitato per la stadiazione e valutazione prognostica, anche se il riscontro di epatite dell’interfaccia e duttopenia sono considerati fattori prognostici negativi. I valori di stiffness epatica (LSM) misurata con VCTE (elastografia transiente) sono strettamente correlati al grado di fibrosi. La LSM è raccomandata non solo per la stratificazione iniziale del rischio, ma anche per la valutazione dinamica della risposta alla terapia e per la individuazione dei pazienti con maggior rischio di evoluzione verso lo scompenso clinico e la morte (4).
La sintomatologia della CBP è variabile e spesso assente per moltissimi anni. Il prurito e la stanchezza rappresentano i più frequenti sintomi all’esordio della malattia, entrambi impattano pesantemente sulla qualità di vita. Il prurito, presente nel 20-80% dei pazienti, di intensità variabile, spesso è l’unico sintomo presente nelle fasi iniziali, e deve indurre il medico al dosaggio di ALP e alla determinazione del profilo autoimmunitario, prima di inviare il paziente a valutazione specialistica.
L’astenia, intesa come stanchezza muscolare, è riferita dal 50-80% dei pazienti, spesso associata a depressione, disturbi cognitivi e del sonno; non è correlata alla severità della CBP.
L’ipercolesterolemia è frequente, causa la comparsa di xantomi e xantelasmi, ma non aumenta il rischio di malattia cardiovascolare e non richiede un trattamento specifico, se non in pazienti con manifestazioni concomitanti della malattia steatosica del fegato correlata ad alterazioni metaboliche (MASLD).
L’osteoporosi è una frequente manifestazione clinica della CBP e necessita adeguata valutazione e terapia.
I pazienti riconosciuti e trattati tardivamente, o che non rispondono alle terapie, mostrano i segni della CE (ittero, epatomegalia, splenomegalia, varici esofagee), e nel tempo presentano le complicanze più gravi (ascite, encefalopatia, emorragia digestiva). Il monitoraggio e trattamento di questi pazienti è sovrapponibile ai casi di CE di altra eziologia. Il rischio di HCC è presente nei pazienti in fase di cirrosi e richiede una sorveglianza ecografica semestrale.
La diagnosi di CBP è spesso facilitata dalla presenza di altre condizioni autoimmuni (sindrome di Sjogren, sclerodermia, malattia di Raynaud, tiroidite, celiachia). Tutti i pazienti con CBP devono ricevere un adeguato screening per queste comorbilità (5).
Aspetti prognostici
La prognosi dei pazienti con CBP è correlata a diversi fattori (età precoce alla diagnosi, sesso maschile, severità della fibrosi alla biopsia, elevati livelli di bilirubina al basale, positività per anti-gp210 o anti-sp100). I pazienti che rispondono alla terapia con acido ursodesossicolico (UDCA) hanno la prognosi migliore.
I due più diffusi modelli prognostici (GLOBE e UK-PBC score) si basano sulla valutazione di alcuni parametri di laboratorio dopo 12 mesi di terapia con UDCA (3).
Più recentemente è stato dimostrato in un’ampia coorte di pazienti che la misurazione della LSM alla diagnosi e il suo andamento durante il trattamento sono correlati alla progressione di malattia, indipendentemente dagli altri fattori prognostici (4). Inoltre, in uno studio retrospettivo su oltre 1000 pazienti è stato dimostrato che la normalizzazione (e non solo la riduzione) di ALP riduce i rischi di scompenso e trapianto, in particolare nei pazienti con LSM ≥ 10 kPa (6). Questi modelli permettono di selezionare i pazienti a maggior rischio che necessitano di iniziare precocemente terapie di seconda linea.
Aspetti terapeutici
 Acidi biliari e derivati
Acidi biliari e derivati
La CBP è considerata una malattia autoimmune, tuttavia il trattamento standard è basato sull’acido ursodesossicolico (UDCA), un acido biliare secondario, con proprietà coleretica, citoprotettiva, anti-infiammatoria e immuno-modulatoria. Alla dose giornaliera di 13-15 mg/kg, l’UDCA ha dimostrato in innumerevoli studi (randomizzati e osservazionali) efficacia in termini di risposta biochimica (riduzione di ALP, GGT, ALT, bilirubina), rallentamento della progressione istologica e miglioramento degli outcome clinici (scompenso, sopravvivenza libera da trapianto, 7), con ottimo profilo di sicurezza, anche in gravidanza. Per tale motivo l’UDCA è indicato in tutte le fasi della malattia (3). In uno studio di coorte internazionale di oltre 3000 pazienti, dopo un anno di terapia, il 40% non presenta riduzione di ALP, ed una parte di essi non tollera il farmaco; una percentuale minore (intorno al 30%) è stata riportata in uno studio italiano di real world (8). UDCA non migliora i principali sintomi (prurito e astenia) che condizionano la qualità di vita della maggior parte dei pazienti con CBP.
Nel 2016 la FDA ha approvato in via condizionata l’acido obeticolico (OCA), in associazione a UDCA, nei pazienti non responder, e in monoterapia nei pazienti non tolleranti ad UDCA. L’OCA, derivato 100 volte più potente dell’acido chenodesossicolico, è un agonista del recettore FXR, regola il metabolismo degli acidi biliari e possiede azione anti-infiammatoria e anti-fibrotica.
Nel trial registrativo (9), e nella analisi a 3 anni, il 46% dei pazienti trattati con 5 mg/die, e il 47% di quelli trattati con 10 mg al giorno, raggiungevano gli endpoint primari dopo 12 mesi di terapia (riduzione di ALP e normalizzazione della bilirubina), rispetto al 10% del placebo. La terapia con OCA determina prurito (che in alcuni casi obbliga all’interruzione del trattamento), ed è controindicata in pazienti con cirrosi scompensata. Numerosi studi di real world hanno confermato l’efficacia di OCA, soprattutto in pazienti non cirrotici; tuttavia l’OCA non ha dimostrato efficacia nel ridurre gli outcome clinici in uno studio randomizzato controllato (RCT) condotto in pazienti con CBP avanzata (10). Nonostante importanti bias presentati dallo studio, nel giugno 2024 l’Agenzia Europea del Farmaco (EMA) ha revocato l’autorizzazione al commercio di OCA nei Paesi Europei, considerando i benefici prodotti dal farmaco minori dei rischi di utilizzo nei pazienti con malattia più avanzata. Dopo una iniziale sospensione della decisione di EMA, dal dicembre 2024 il farmaco non è più prescrivibile in Italia e il trattamento deve essere sospeso.
Agonisti di PPAR
Il bezafibrato (agonista pan-PPAR) e il fenofibrato (agonista PPARα) sono stati valutati in pazienti con risposta parziale o non responder a UDCA, con efficacia in termini di risposta biochimica e di riduzione dei sintomi (prurito, astenia); tuttavia il loro utilizzo è off label.
Infine, nel 2024 FDA ha approvato altri due agonisti di PPAR in monoterapia o in combinazione ad UDCA per il trattamento dei pazienti con risposta inadeguata ad UDCA. Gli agonisti di PPAR modulano il metabolismo degli acidi biliari, hanno azione anti-infiammatoria e anti-fibrotica, regolano il metabolismo lipidico, migliorano la sensibilità insulinica.
Elafibranor (agonista di PPAR-α e δ), alla dose di 80 mg/die per 12 mesi, ha dimostrato efficacia nel 51% dei pazienti (vs 4% del placebo), con un buon profilo di sicurezza (11).
Seladelpar (agonista di PPAR-δ), alla dose di 10 mg/die per 12 mesi, ha raggiunto l’endpoint primario (riduzione di ALP e normalizzazione di bilirubina) nel 61.7% dei pazienti (vs 20% del placebo, 12).
Entrambi non devono essere utilizzati in pazienti con cirrosi scompensata.
Elafibranor e Seladelpar, in attesa di autorizzazione da parte delle Autorità regolatorie, sono disponibili in Italia, nei Centri di riferimento, nell’ambito di programmi di uso compassionevole, in pazienti non responsivi o intolleranti a UDCA.
Un aspetto importante del management della CBP è il trattamento dei sintomi che condizionano la qualità di vita dei pazienti. I farmaci sinora disponibili per il prurito, quali colestiramina, rifampicina, naltrexone, non hanno dimostrato efficacia. Il bezafibrato è risultato più efficace del placebo in un RCT (13). In prospettiva futura sembrano promettenti i risultati di seladelpar e di una nuova classe di farmaci, gli inibitori del trasporto ileale degli acidi biliari (IBAT), autorizzati nel trattamento del prurito delle malattie colestatiche genetiche. Non esistono in atto cure efficaci per la stanchezza e i disturbi cognitivi, che rappresentano un pesante burden di malattia, anche se alcuni farmaci in fase 2 di sperimentazione sembrano promettenti.
Conclusioni
La CBP è una malattia rara ma con pesante impatto in termini di morbilità e mortalità nei pazienti trattati tardivamente o non responsivi alle cure. La prospettiva terapeutica è cambiata profondamente negli ultimi anni. Numerosi farmaci efficaci sono già disponibili e altri lo saranno in un prossimo futuro, rendendo possibile il rallentamento della malattia e la prevenzione delle complicanze più gravi.
Poiché le cure attuali non sono adeguate per i pazienti con cirrosi in fase avanzata non trapiantabili, appare evidente la necessità di attuare strategie di diagnosi e trattamento precoci che permettano di rispondere efficacemente alle necessità dei pazienti.
- Colapietro F, Bertazzoni A, Lleo A. Contemporary epidemiology of primary biliary cholangitis. Clin Liv Dis 2022;26(4):555-570.
- Marzioni M, Bassanelli C, Ripellino C, et al. Epidemiology of primary biliary cholangitis in Italy: evidence from a real-world database. Dig Liv Dis 2018;51:724-729.
- Lindor KD, Bowlus CL, Boyer J, et al. Primary biliary cholangitis: 2018 practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2019,69(1):394-419.
- Lam L, Soret PA, Lemoinne S, et al. Dynamics of liver stiffness measurement and clinical course of primary biliary cholangitis. Clin Gastroenterol Hepatol 2024; doi:10.1016/j.cgh.2024.06.035.
- Carbone M, Gerussi A, Cardinale V, AISF Expert Panel. Position paper of the Italian Association for the Study of the Liver (AISF): management and treatment of primary biliary cholangitis. Dig Liver Dis 2024;56:1461-1474.
- Corpechot C, Lemoinne S, Soret PA, et al. Adequate versus deep response to ursodeoxicholic acid in primary biliary cholangitis: to what extent and under what conditions is normal alkaline phosphatase level associated with complication-free survival gain? Hepatology 2024;79(1):39-48.
- Harms SH, Van Buuren HR, Corpechot C, et al. Ursodeoxicholic acid therapy and liver transplant-free survival in patients with primary biliary cholangitis. J Hepatol 2019;71:357-365.
- Vespasiani-Gentilucci U, Rosina F, Pace-Palitti V, et al. Rate of non-response to ursodeoxicholic acid in a large real-world cohort of primary biliary cholangitis patients in Italy. Scand J Gastroenterol 2019;54(10):1274-1282.
- Nevens F, Andreone P, Mazzella G, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 2016;375:631-643.
- Kowdley KV, Hirschfield GM, Coombs C, et al. COBALT: a confirmatory trial of obethicolic acid in primary biliary cholangitis with placebo and external controls. Am J Gastroenterol 2024; doi: 10.14309/ajg.0000000000003029. Online ahead of print.
- Kowdley KV, Bowlus CL, Levy C, et al. Efficacy and safety of Elafibranor in primary biliary cholangitis. N Engl J Med 2024;390:795-805.
- Hirschfield GM, Bowlus CL, Mayo MJ, et al. A phase 3 trial of Seladelpar in primary biliary cholangitis. N Engl J Med 2024;390:783-794.
- De Vries E, Bolier R, Goet J, et al. Fibrates for itch (FITCH) in fibrosing cholangiopathies: a double-blind, randomized placebo-controlled trial. Gastroenterology 2021;160:734-743.
